The multiMiR R package and database: integration of microRNA–target interactions along with their disease and drug associations by Yuanbin Ru, et al. ( Nucl. Acids Res. (2014) doi: 10.1093/nar/gku631)
Abstract:
microRNAs (miRNAs) regulate expression by promoting degradation or repressing translation of target transcripts. miRNA target sites have been catalogued in databases based on experimental validation and computational prediction using various algorithms. Several online resources provide collections of multiple databases but need to be imported into other software, such as R, for processing, tabulation, graphing and computation. Currently available miRNA target site packages in R are limited in the number of databases, types of databases and flexibility. We present multiMiR, a new miRNA–target interaction R package and database, which includes several novel features not available in existing R packages: (i) compilation of nearly 50 million records in human and mouse from 14 different databases, more than any other collection; (ii) expansion of databases to those based on disease annotation and drug microRNAresponse, in addition to many experimental and computational databases; and (iii) user-defined cutoffs for predicted binding strength to provide the most confident selection. Case studies are reported on various biomedical applications including mouse models of alcohol consumption, studies of chronic obstructive pulmonary disease in human subjects, and human cell line models of bladder cancer metastasis. We also demonstrate how multiMiR was used to generate testable hypotheses that were pursued experimentally.
Amazing what you can do with R and a MySQL database!
The authors briefly describe their “cleaning” process for the consolidation of these databases on page 2 but then note on page 4:
For many of the databases, the links are available. However, in Supplementary Table S2 we have listed the databases where links may be broken due to outdated identifiers in those databases. We also listed the databases that do not have the option to search by miR NA-gene pairs.
Perhaps due to editing standards (available for free lance work) I have allergy to terms like “many,” especially when it is possible to enumerate the “many.”
In this particular case, you have to download and consult Supplementary Table S2, which reads:
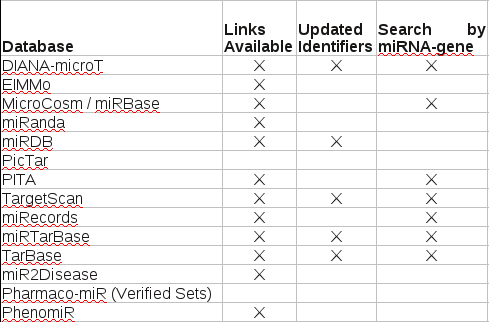
The explanation for this table reads:
For each database, the columns indicate whether external links are available to include as part of multiMiR, whether those databases use identifiers that are updated and whether the links are based on miRNA-gene pairs. For those database that do not have updated identifiers, some links may be broken. For the other databases, where you can only search by miRNA or gene but not pairs, the links are provided by gene, except for ElMMo which is by miRNA because of its database structure.
Counting I see ten (10) databases with a blank under “Undated Identifiers” or Search by miRNA-gene,” or both.
I guess ten (10) out of fourteen (14) qualifies as “many,” but saying seventy-one percent (71%) of the databases in this study lack either “Updated Identifiers,” “Search by miRNA-gene,” or both, would have been more informative.
Potential records with these issues? EIMMo, version 4 has human (50M) and mouse (15M), MicroCosm / miRBase human (879054), and miRanda (assuming human, Good mirSVR score, Conserved miRNA), 1097069. For the rest you can consult Supplemental Table 1, which lists URLs for the databases and dates of access, but where multiple human options are available, not which one(s) were selected.
The number of records for each database that may have these problems also merits mention in the description of the data.
I can’t comment on the usefulness of this R package for exploring the data but the condition of the data it explores needs more prominent mention.
